Algorithmique moléculaire pour la conception de médicaments

L'émergence de méthodes algorithmiques en biologie moléculaire remonte sans doute aux années 80, mais le processus s'est accéléré depuis peu comme en témoigent la création de journaux spécifiques, la mise en place de programmes pluri-disciplinaires de recherche, etc. De façon schématique, ces activités sont de deux ordres.
Les premières sont liées à l'étude du génome. D'un point de vue physico-chimique, cela consiste à proposer des modèles de stabilité des acides nucléiques ---ARN et ADN--- et des protéines. Ces modèles sont utilisés pour prédire la stabilité des molécules étudiées, e.g. les structures secondaires et tertiaire des protéines. D'un point de vue combinatoire, il s'agit de comparer des séquences génétiques et de modéliser les dérives phylogenétiques associées.
Les secondes sont liées à la conception de médicaments et visent à trouver plus rapidement des molécules plus efficaces, moins nocives, et plus faciles à extraire ou à synthétiser. Les questions posées sont souvent de nature géométrique et/ou combinatoire, d'où l'intérêt suscite dans le projet Prisme. Les questions majeures de la conception de médicaments peuvent être décrites comme suit.
|
Améliorer ou créer un médicament peut se faire soit en concevant une
nouvelle molécule ex nihilo, soit en remplaçant un (plusieurs)
groupement(s) chimique(s) sur une structure déjà connue. L'objet de la
synthèse combinatoire est précisément d'explorer systématiquement tous
ces possibles. Cette exploration peut être réelle comme indique sur
l'image ci-contre. Elle peut aussi être virtuelle
i.e. informatique. De façon imagée, les molécules sont alors vues
comme des phrases et toutes les phrases utilisant certaines règles
grammaticales et un certain vocabulaire sont examinées.
Les difficultés de l'approche sont multiples: identifier rapidement celles des molécules ainsi générées qui sont stables, prédire l'activité des molécules, repérer les molécules similaires, etc. |
 |
| Synthèse combinatoire |
|---|
|
Les méthodes récentes de synthèse moléculaire ---e.g. la synthèse
combinatoire--- et/ou les concentrations horizontales d'entreprises
pharmaceutiques font que les chimistes sont souvent confrontés à de
grandes bases de données moléculaires. Fusionner de telles bases de
données, les organiser en groupes de molécules similaires sont des
étapes nécessaires pour les rendre plus facilement exploitables. Cela
permet par exemple en phase d'exploration fonctionnelle, de tester
systématiquement tous les membres d'un groupe après que l'un d'entre
eux pris au hasard a révélé un comportement chimique intéressant.
Les difficultés rencontrées sont de deux ordres. D'une part, étant donné le codage des molécules communément utilisé, les problèmes a résoudre sont formules dans des espaces de grande dimension. D'autre part, les problèmes de clustering sont généralement NP-hard. |
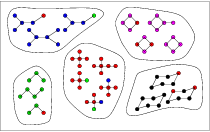 |
| Clustering |
|---|
 |
Les degrés de liberté de rotation associés aux liaisons covalentes,
ainsi que les forces d'attraction-répulsion d'origine électro-statique
principalement, sont tels que certaines conformations moléculaires
sont plus stables que d'autres. L'objectif est donc la recherche de
ces minima d'énergie parallèlement à l'étude expérimentale par des
rayons X ou la résonance magnétique nucléaire.
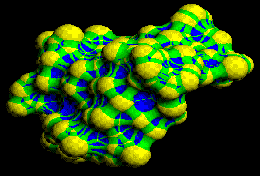
D'un point de vue algorithmique, la recherche de ces états stables est un problème de minimisation non linéaire où le nombre de variables est le nombre de degrés de liberté de la molécule. |
| Surface moléculaire |
|---|
| Les mécanismes hormonaux de communication cellulaire , la réponse immunitaire ou encore l'action des médicaments font intervenir la fixation d'une molécule ---neuro-transmetteur, anti-gène ou médicament--- sur des récepteur des membranes cellulaires. Cette fixation ou docking requiert une complémentarité géométrique ainsi qu'une interaction électro-statique favorable. Son étude est une étape clef de la conception de médicaments et nécessite des modèles deformables de surfaces moléculaires. | 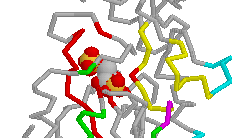 |
| Site actif enzymatique |
|---|
| Exemple de surface moleculaire |
|---|
|
Lorsque plusieurs molécules sont connues pour avoir le même effet, une
question consiste à trouver les groupes d'atomes responsables de cette
activité. Ces groupes atomes appelés pharmacophorespeuvent
être les mêmes, mais ils peuvent aussi différer, le point commun étant
alors la création d'un champ électro-statique identique permettant la
fixation du ligand.
Les méthodes de recherche de pharmacophores devraient en théorie tenir compte de la dynamicité des surfaces moléculaires. En pratique, elles opèrent sur des modèles moléculaires figes. L'une d'entre elles consiste à chercher e.g. des triplets d'atomes à égale distance dans les molécules étudiées. Une autre technique utilisée par les chimistes réside dans l'examen des formules développées des molécules. Si ces dessins sont normalisés, l'oeil averti arrive à identifier les groupements chimiques d'intérêt. |
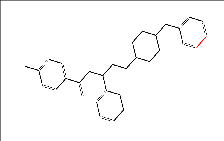 |
| Dessin de molécule |
|---|
| Last modified: Fri Nov 19 09:47:17 MET 1999 - Frederic.Cazals@sophia.inria.fr | F. Cazals | Thèmes |
|